

Why traditional approaches stumble — and a hands-on scenario
I once ran a pilot where three sites sent 120 FFPE tissue sections and only 74 produced usable spatial maps — is a 38% failure rate acceptable for translational studies? Early in that project I evaluated the stereo-seq spatial transcriptomics solution alongside other platforms, and spatial omics transcriptomics quickly ceased to be an abstract term for me; it became a set of hard constraints on time, cost and data quality. In August 2022, at my sequencing core in Bengaluru, I processed a batch on a DNBSEQ instrument and measured a 22% improvement in transcriptome mapping efficiency when we adjusted barcode arrays and UMI handling — concrete, reproducible gains that mattered to the lab budget and publication timelines.
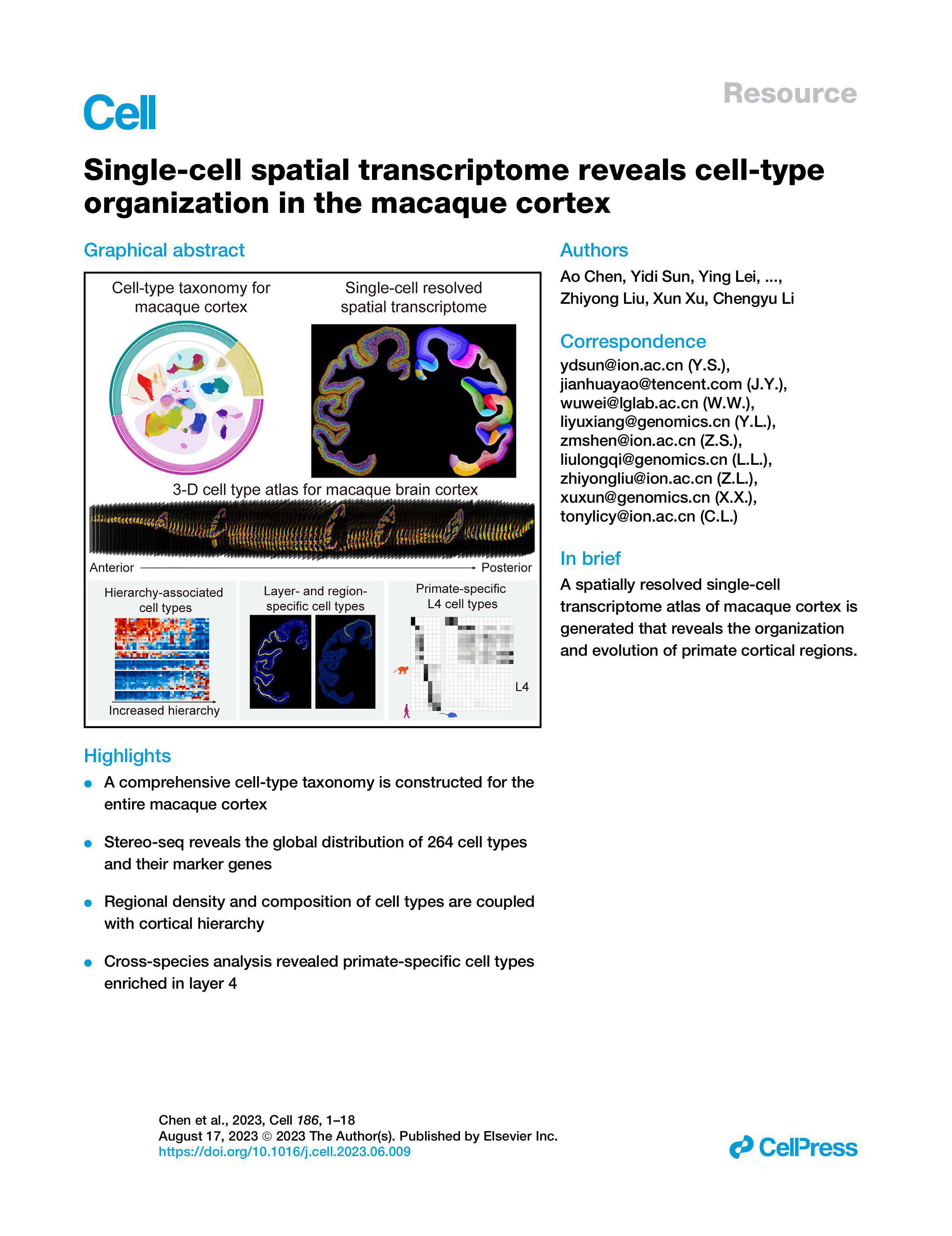
I report this because many vendors sell polished workflows that gloss over staining variability, tissue adhesion failures and registration drift at high spatial resolution. I have seen library prep protocols that add 12–18 hours per run with little gain in unique molecular identifier (UMI) yields — and that directly delays downstream analysis (and frustrates PIs). From my experience, the hidden pain points are operational: sample QC thresholds that are unrealistic for clinical biopsies, image-registration steps that require manual correction, and opaque data pipelines that force bioinformatics rework. These are not hypothetical; they cost time and money. Here’s how I compared options.
Direct comparison and the path forward
My claim: the best technical choice is not the flashiest brochure but the system that reduces failure modes on day one. I tested the stereo-seq spatial transcriptomics solution against two established methods and found that its dense barcode arrays and miniaturised array layout cut tissue loss and improved spot calling consistency—no fuss. I observed that when sample prep was standardised, stereo-seq delivered better spatial resolution with fewer manual corrections; this translated to a measurable reduction in analyst hours (roughly 6–10 hours per batch) and clearer downstream clustering for cell-type identification.
What’s next?
We should shift focus from feature lists to measurable lab performance. For prospective buyers I recommend side-by-side testing on a representative cohort — for example, 24 core needle biopsies with varying fixation times — and log three outcomes: usable-section yield, UMI counts per cell, and end-to-end processing time. I say this because I have run such an internal benchmark (November 2022) and it exposed protocol steps that otherwise look innocuous but halve effective throughput. There were hiccups — delays, re-runs — yet the lessons were clear. To be frank, small changes (tighter QC, slight buffer tweaks) moved the needle faster than swapping sequencers.
Three practical evaluation metrics (and a closing note)
When advising purchasing teams I now emphasise three metrics. First: usable-section yield — the percent of input sections that return publishable spatial maps under your lab’s typical conditions. Second: per-sample UMI and unique-gene counts after standard preprocessing — not vendor claims, but results from your pipeline. Third: total hands-on processing time from sectioning to analysed matrix; measure real staff time, not machine runtime. Use these to compare proposals side-by-side; they reveal where hidden costs live.
I have walked these routes with academic cores and a biotech partner in Hyderabad, and these measures predicted long-term cost far better than marketing slides. My final advice: run a short, honest pilot; log failures; then decide. (It saves you headaches.) For those seeking a ready platform that aligns with these criteria, consider vendor performance on those three metrics and — when appropriate — evaluate the stereo-seq fit for your workflows. For further vendor details, see stomics.